BioEmu: A Next-Generation AI System for Protein Flexibility Modelling
BioEmu:
Context:
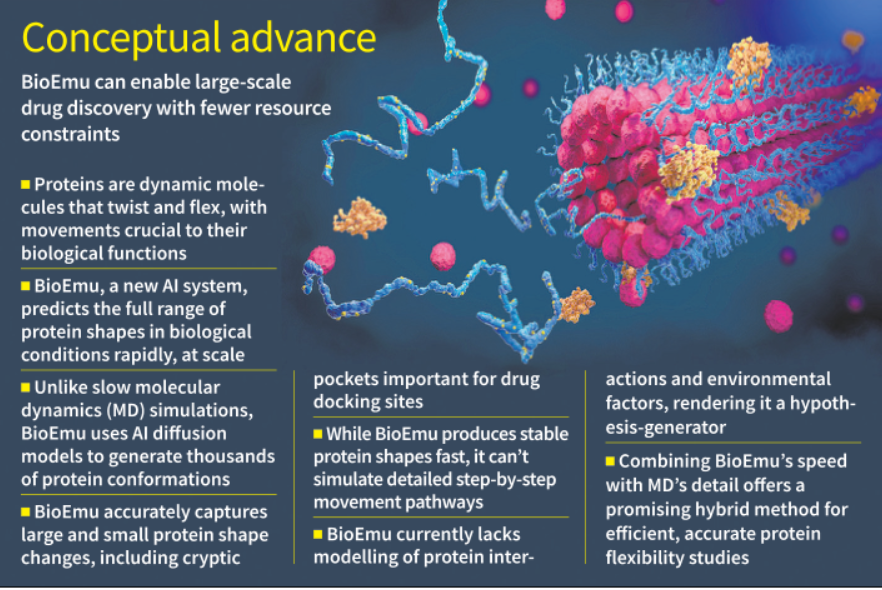
BioEmu is a cutting-edge deep learning system developed by Microsoft, Rice University (USA), and Freie Universität (Germany), designed to predict protein flexibility at scale, surpassing traditional methods like Molecular Dynamics (MD) in speed and cost-efficiency.
Why Protein Flexibility Matters:
-
Proteins are dynamic molecules; their functions depend on shape changes (e.g., enzymes opening, signalling proteins shifting).
-
Existing tools like AlphaFold predict static structures, often failing to capture real-time conformational diversity.
-
Flexibility modeling is crucial for understanding drug binding, protein switching, and mutation effects.
How BioEmu Works:
-
BioEmu uses a diffusion-based AI model to simulate the equilibrium ensemble — all biologically relevant conformations of a protein.
-
Trained on:
-
Millions of AlphaFold-predicted structures
-
200 ms worth of MD simulations of thousands of proteins
-
~5 lakh experimental mutant sequences
-
-
Working Principle:
Like reconstructing a sugar cube from scattered particles, BioEmu learns to generate stable protein shapes from noise.
What is an Equilibrium Ensemble?
Think of a protein or molecule as something that doesn’t stay frozen in one shape — it constantly moves and wiggles, especially at body temperature.
Now imagine you take a snapshot of the protein every second over a long time. Each snapshot shows the different shapes (also called conformations) the protein takes. These shapes are not random — some are more likely than others, depending on energy and stability.
An equilibrium ensemble is:
A collection of all the possible stable shapes a molecule can take under constant temperature and pressure, when it’s in a balanced (equilibrium) state.
Advantages Over Classical MD:
| Feature | BioEmu | Molecular Dynamics (MD) |
|---|---|---|
| Speed | Minutes to hours on a GPU | Tens of thousands of GPU-hours |
| Cost | Low | High |
| Scale | Thousands of proteins | Limited |
| Output | Ensemble of stable shapes | Time-based movement pathways |
-
Accurate in predicting:
-
Large conformational shifts (83%)
-
Local unfolding and cryptic drug-binding pockets
-
Structural instability due to mutations
-
Limitations:
-
No time-evolution modeling (can’t show step-by-step protein folding like MD).
-
Cannot simulate:
-
Temperature shifts, pH changes
-
Interactions with drugs or other proteins
-
Membranes or multi-chain complexes
-
-
No confidence score (unlike AlphaFold)
BioEmu vs Molecular Dynamics (MD)
| Component | BioEmu | Molecular Dynamics (MD) |
|---|---|---|
| Main Function | Predicts the range of stable protein shapes | Simulates step-by-step motion of atoms |
| Output | Static snapshots of all likely conformations | Dynamic transitions between states |
| Time Scale | Minutes to hours (on a single GPU) | Days to weeks (on supercomputers) |
| Resource Requirement | Low | High |
| Predicts Pathways? | ❌ No – Does not show how protein changes form | ✅ Yes – Shows transition mechanisms |
| Handles Environment (e.g. water, drugs, pH)? | ❌ No | ✅ Yes |
| Protein Interactions? | ❌ Only single chains | ✅ Multi-protein and complex environments |
| Best Use Case | Hypothesis generation at scale | Detailed mechanism and reliability analysis |
Applications and Implications:
-
Drug Discovery: Rapid identification of hidden binding pockets across protein databases.
-
Biomedical Research: Understanding mutation-driven diseases like cancer (e.g., Ras protein modeling).
-
Computational Biology: Enables hypothesis generation at scale with limited computing resources.